- 首页 > 正文
深三院结核之窗丨结核分枝杆菌“隐身术”被破解?新研究揭示免疫逃逸关键通路
感染医线 发表时间:2025/12/15 19:23:50
编者按:在结核病治疗中,仍有部分患者面临传统疗法效果有限的困境。近年研究逐渐揭示,结核菌可巧妙干扰宿主免疫应答,延迟T细胞抵达感染部位,从而逃避清除。在本期“结核之窗”栏目中,卢水华教授团队分享了一项研究,深入探索了结核菌通过色氨酸代谢途径影响免疫细胞功能的机制,为难治性结核的干预提供了新视角。该工作不仅深化了对结核免疫逃逸的认识,也为发展宿主导向的免疫治疗策略带来了希望。
研究简介
一、研究背景
结核分枝杆菌(Mtb)是导致结核病的病原体,可以操纵宿主反应以延迟T细胞向肺部的募集,从而延迟适应性免疫的发生。这种延迟被广泛认为是Mtb建立其生态位的主要策略,并且可能是通过适应性免疫根除Mtb的关键瓶颈。先前的代谢组学分析表明,Mtb感染会诱导巨噬细胞中色氨酸(Trp)代谢增加。Trp代谢物犬尿氨酸(Kyn)是炎症和T细胞活性的有效负调节剂。吲哚胺2,3-双加氧酶(IDO1)是一种参与Trp代谢的酶,能够将Trp转化为Kyn。在免疫调节中,IDO1的活化可以导致局部Trp的耗竭,从而抑制T细胞的活性,这是许多病原体逃避免疫反应的策略之一。
芳香烃受体(AhR)是一种转录因子,参与多种生物学过程,包括细胞分化、免疫反应和代谢调控。AhR的活化可以通过多种机制影响免疫细胞的功能,包括调节T细胞的分化和活性,通过Treg-巨噬细胞抑制轴驱动T细胞功能障碍。然而,目前尚不清楚Kyn是否可以影响炎症巨噬细胞招募T细胞,从而影响结核病的适应性免疫反应。
本研究发现Mtb通过增加炎症巨噬细胞中IDO1的表达来促进Kyn的产生,从而增加细胞内AhR的表达。AhR随后上调细胞因子信号传导抑制因子3(SOCS3)的表达,从而抑制JAK-STAT1通路的激活。这导致趋化因子表达减少,从而损害T细胞向肺部感染部位的迁移。
研究方法与结果
1. 高水平的Kyn会抑制肺部结核患者体内T细胞的浸润
结核患者血清Kyn水平显著升高(图1A)。为了评估Kyn对CD4+和CD8+T细胞功能的影响,将结核病患者分为高Kyn表达组和低Kyn表达组(图1B)。与Kyn低表达患者相比,血清中Kyn高表达患者的肺组织浸润CD4+和CD8+T细胞数量显著减少(图1C、D)。患者血清中的Kyn水平与T细胞表达效应细胞因子IFN-γ和TNF-α之间呈负相关(图1E)。Kyn水平与外周血T细胞中抑制性受体(包括PD-1、TIM3和LAG3)的表达水平呈正相关(图1F)。Mtb诱导宿主Trp代谢产生大量代谢物Kyn,从而影响肺内效应T细胞的浸润水平和功能,实现免疫逃避。
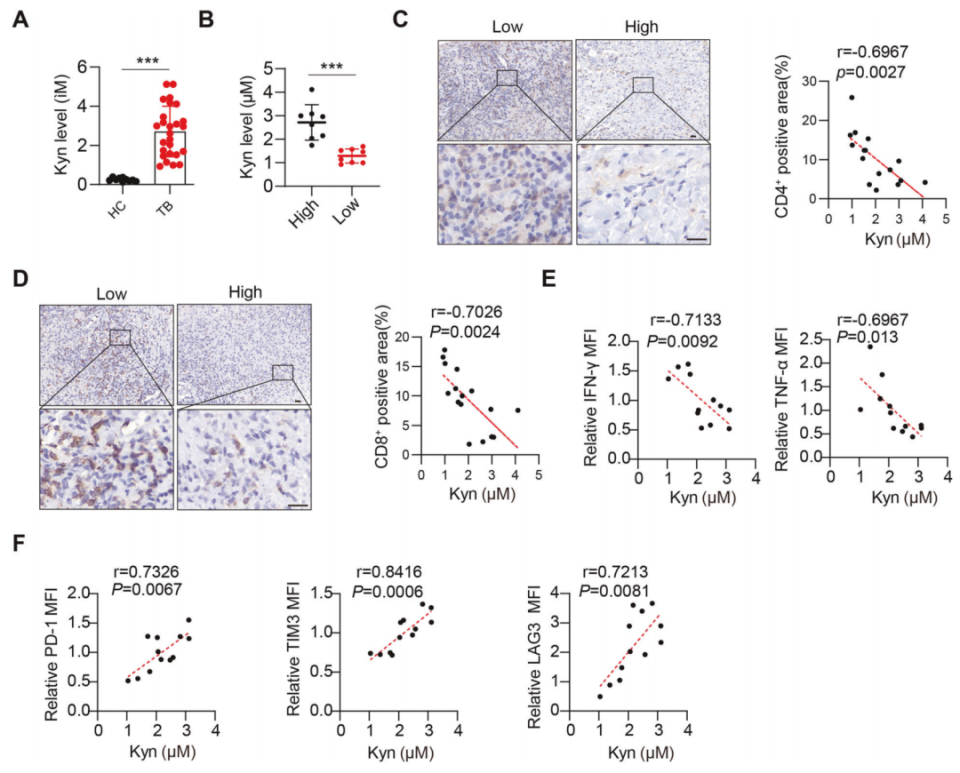
图1. Kyn抑制结核病患者的T细胞浸润
2. Mtb通过增强炎症巨噬细胞的IDO1-Kyn代谢途径来抑制T细胞的浸润
感染Mtb后,炎症巨噬细胞中IDO1的表达水平显著增加,其他色氨酸降解酶,如IDO2和TDO2,没有显著变化(图2A)。作者首先研究了巨噬细胞中IDO1的表达水平是否以Mtb负荷依赖性方式诱导。结果显示,IDO1水平随着Mtb感染复数(MOI)和感染持续时间的增加而增加(图2B-D)。与未感染小鼠相比,Mtb感染小鼠的肺组织中IDO1的表达也增加(图2E),特别是在肺泡巨噬细胞中(图2F)。
既往研究报道结肠癌细胞可以通过增加IDO1mRNA的稳定性来增加IDO1的表达。Mtb感染细胞中IDO1mRNA的降解率显著低于未感染细胞(图2G)。研究者使用LC-MS检测了Mtb感染前后巨噬细胞内Kyn的水平,发现Mtb感染导致IDO1显著上调,同时细胞内Kyn产量大幅增加(图2H)。结核病患者肺部IDO1mRNA的表达水平与CD4+T和CD8+T细胞的浸润呈负相关(图2I)。
体外T细胞迁移测定证实,与对照组相比,敲低炎症巨噬细胞中的IDO1增加了CD4+和CD8+T细胞的迁移(图2J)。相反,IDO1的过表达则抑制了CD4+和CD8+T细胞的迁移(图2K)。为了进一步探究IDO1是否通过从Trp代谢中产生Kyn来影响T细胞迁移,作者在培养基中添加了额外的Kyn。有趣的是,作者发现Kyn逆转了IDO1敲低引起的CD4+和CD8+T细胞的迁移(图2L)。同样,在缺乏Trp的条件下,炎症巨噬细胞在体外促进了CD4+和CD8+T细胞的迁移;然而,通过额外补充Kyn,这种现象可以被消除(图2M)。
敲低IDO1显著增加了炎症巨噬细胞中CXCL9和CXCL10的表达水平,而其他趋化因子则没有显著变化(图2N)。当炎症巨噬细胞中IDO1过度表达时,CXCL9和CXCL10的表达水平下降(图2O-P)。在炎症巨噬细胞中用针对CXCL9和CXCL10的中和抗体进行治疗,可以抵消IDO1敲低后的T细胞迁移增强(图2Q)。在IDO1缺陷的炎症巨噬细胞中,添加Kyn导致CXCL9/10的mRNA水平降低(图2R)。ELISA进一步证实,敲除IDO1,然后补充Kyn可降低CXCL9和CXCL10的蛋白水平(图2S)。在色氨酸缺乏的条件下培养的炎症巨噬细胞呈现出CXCL9/10表达水平升高;通过额外补充Kyn可以逆转这种现象(图2T)。
这些结果表明,IDO1通过将Trp代谢为Kyn来抑制CXCL9和CXCL10的产生,从而抑制T细胞迁移。
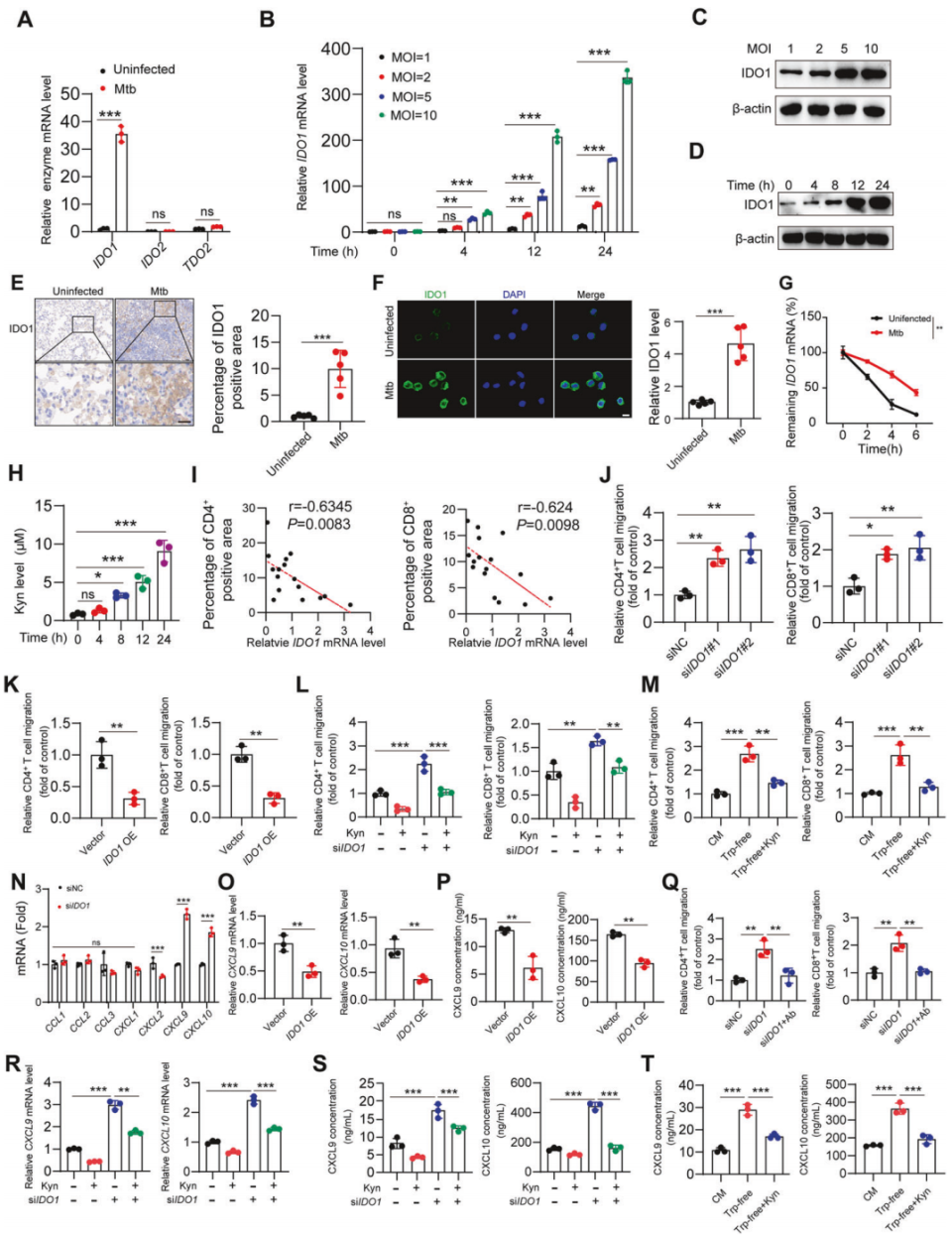
图2. Mtb通过激活IDO1-Kyn通路抑制T细胞浸润
3. IDO1通过STAT1-CXCL9/10信号通路抑制T细胞浸润
STAT1、NF-κB和CREB(腺苷3′,5′-单磷酸反应元件结合蛋白)结合蛋白(CBP)是介导CXCL9和CXCL10转录的转录因子。敲低STAT1,而不是NF-κB或CBP,可以消除IDO1敲低引起的CXCL9和CXCL10蛋白水平升高(图3A、B)。qPCR实验还证实,敲低STAT1消除了IDO1敲低引起的CXCL9和CXCL10mRNA水平的增加(图3C)。在炎症巨噬细胞中敲低IDO1后,STAT1的基线丰度保持不变,但STAT1酪氨酸磷酸化增加。添加Kyn后,这种效应发生逆转(图3D)。在Trp缺乏的条件下,观察到与IDO1敲低相似的结果(图3E)。
综上,Mtb增强IDO1的表达,并利用其代谢物Kyn抑制STAT1-CXCL9/10信号通路。
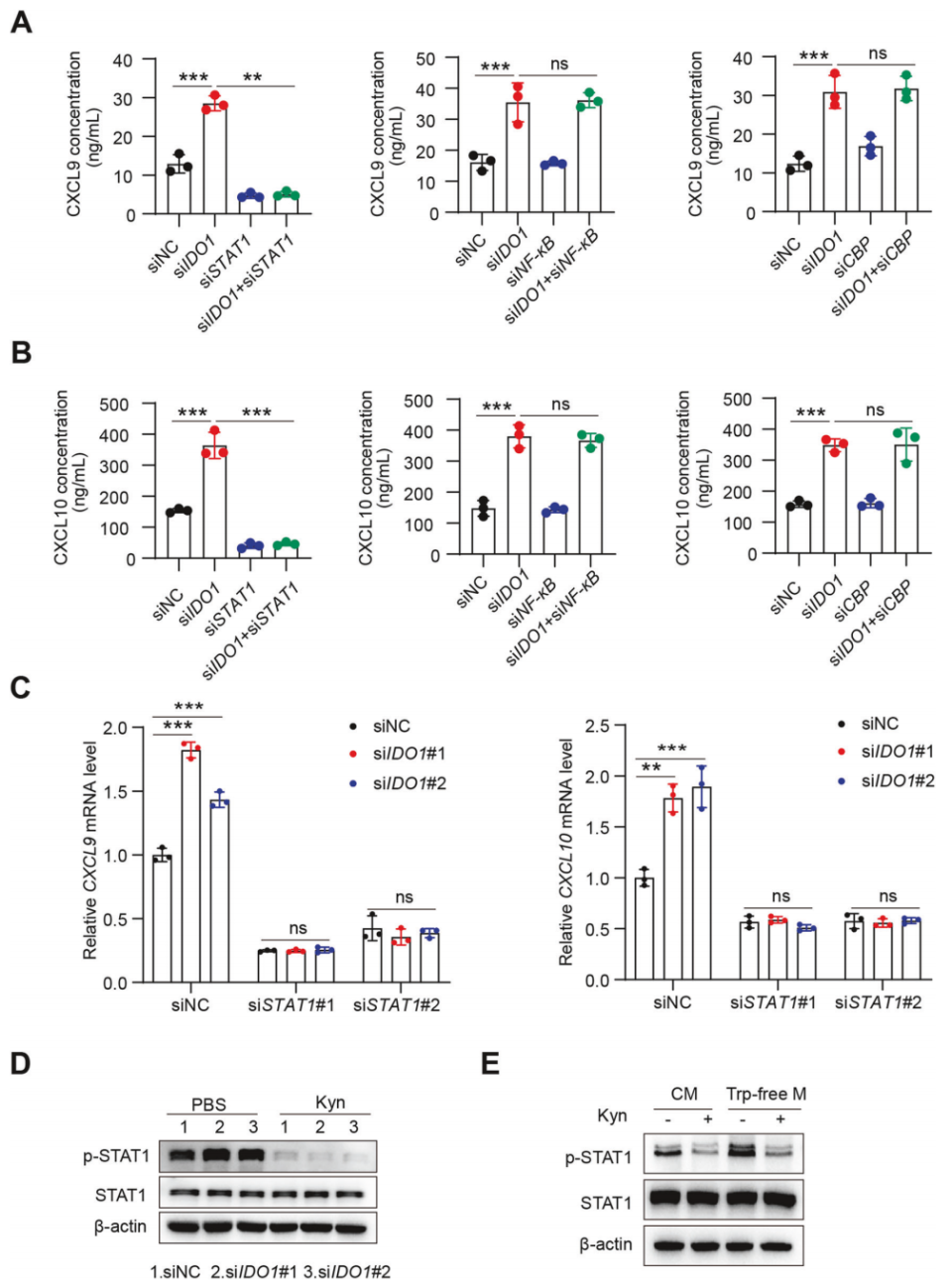
图3. Mtb通过抑制STAT1通路下调CXCL9/10表达
4. IDO1-Kyn代谢途径通过AhR的激活来抑制STAT1的磷酸化过程
先前的研究表明Kyn是AhR的内源配体。配体结合后,AhR易位到细胞核中并上调两个AhR靶基因的表达,即编码细胞色素P450家族1亚家族A多肽1(CYP1A1)和细胞色素P450家族1亚家族B多肽1(CYP1B1)的基因,以发挥其生物学效应。与未感染的炎症细胞相比,AhR、CYP1A1和CYP1B1的表达在Mtb感染的炎症巨噬细胞中显著上调(图4A-C)。免疫荧光实验的结果表明,Mtb感染后,AhR从细胞质转移到细胞核(图4D)。作者试图阐明AhR是否参与Trp-Kyn代谢途径来调节炎症巨噬细胞对T细胞的趋化性。作者发现AhR的敲低促进了炎症巨噬细胞对T细胞的趋化作用(图4E、F)。添加外源性Kyn无法消除这种趋化作用(图4G)。
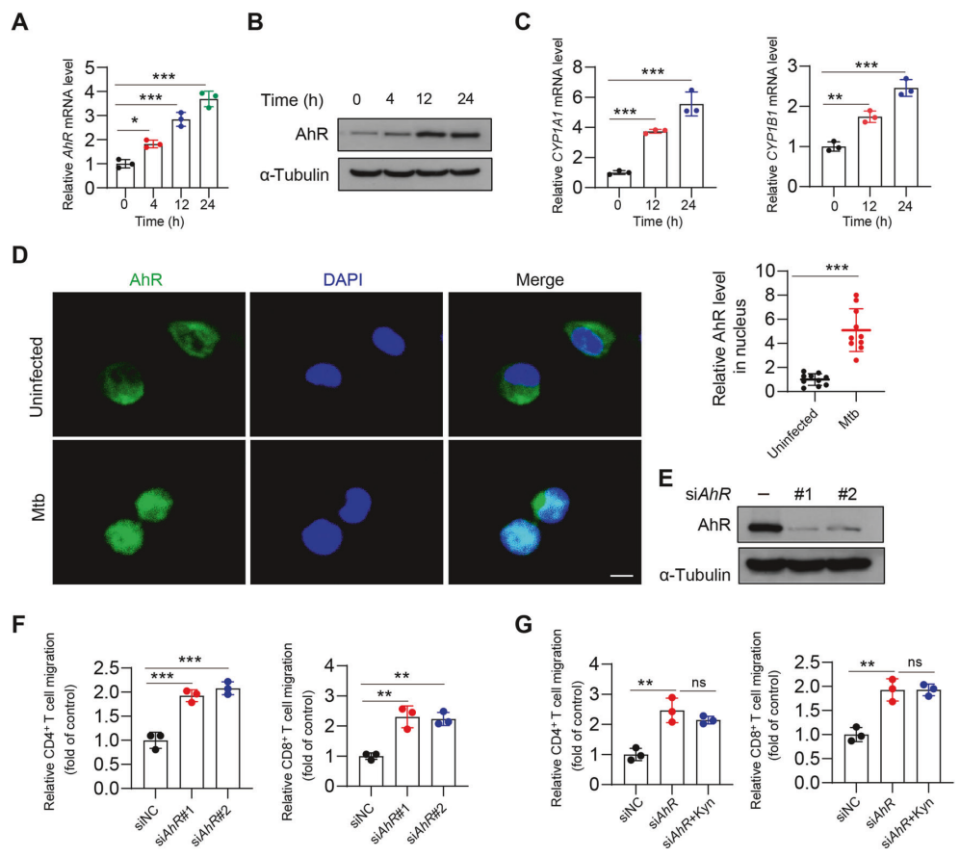
图4. Mtb激活AhR以抑制T细胞迁移
5. Mtb激活AhR蛋白从而抑制STAT1-CXCL9/10通路。
为了进一步探讨AhR对STAT1−CXCL9/10通路的影响,观察到在AhR抑制后,炎症巨噬细胞的CXCL9和CXCL10水平显著升高(图5A、B)。然而,添加外源Kyn并不影响CXCL9/10的水平(图5C、D)。在炎症性巨噬细胞中,STAT1的敲除改善了AhR诱导的CXCL9和CXCL10的mRNA水平的增加(图5E、F)。敲除AhR或使用AhR抑制剂SR-1也促进了STAT1的磷酸化;然而,添加外源性Kyn加入无法抑制磷酸化的STAT1(图5G、H)。综上,IDO1-Kyn代谢途径激活AhR,导致STAT1激活及其下游效应分子表达的抑制。
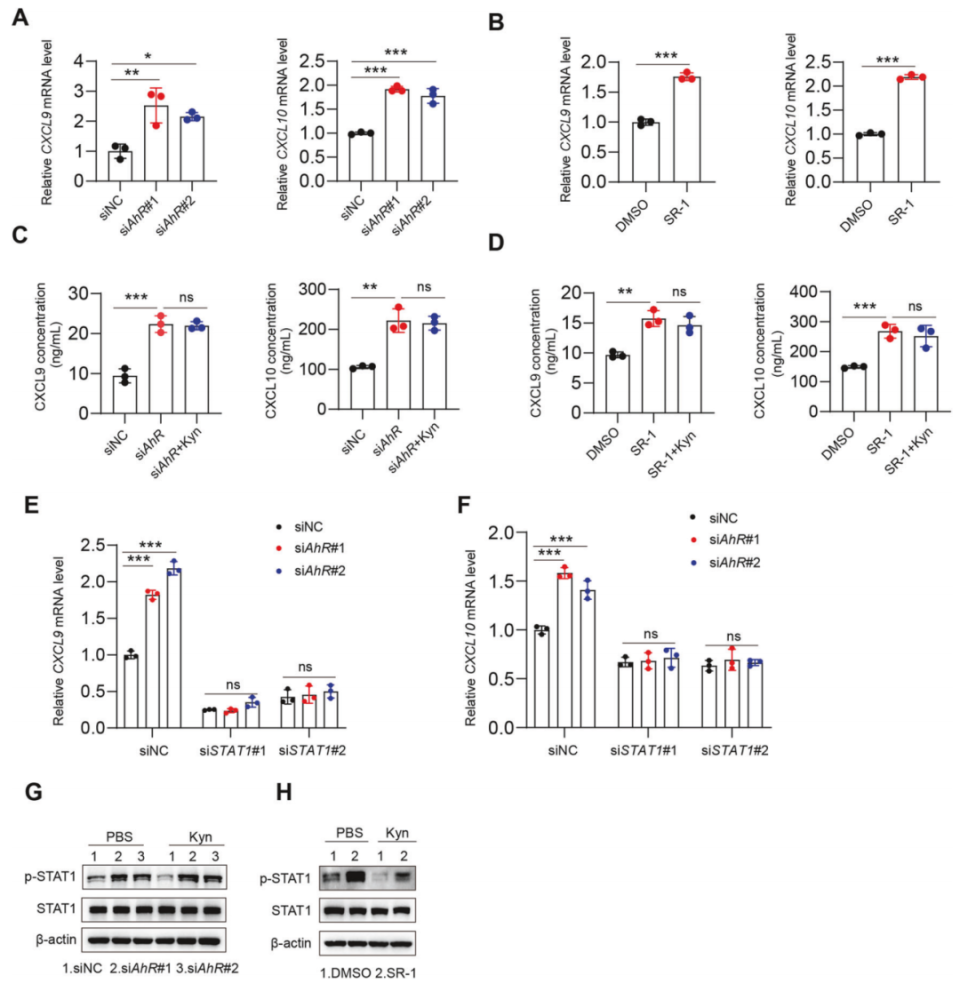
图5. Mtb激活AhR以减弱STAT1-CXCL9/10通路
6. AhR通过上调SOCS3的表达,减少了JAK-STAT1的磷酸化过程
SOCS作为JAK-STAT通路的直接负调节因子,包括SOCS1-7和CISH。在巨噬细胞中敲低AhR后,只有SOCS3mRNA表达水平显著下调(图6A)。作者假设AhR可能会触发巨噬细胞中SOCS3的表达以响应Mtb感染。作者进一步发现SOCS3启动子包含AhR结合核心序列(5'-GCGTG),如JASPAR数据库所示。ChIP-qPCR和CUT&Tag分析的结果表明,Kyn处理组中AhR与SOCS3启动子的结合比PBS或IgG处理组更强(图6B、C)。
双荧光素酶报告基因测定表明,AhR与SOCS3启动子的结合增加了报告基因的表达(图6D)。当SOCS3启动子中的5'-GCGTG核心序列突变为5'-GCATG时,这种效应被消除(图6D)。随着时间的增加和Mtb数量的增加,SOCS3的表达水平也增加(图6E,F)。
此外,当通过siRNA敲低SOCS3时(图6G),炎症巨噬细胞表现出T细胞迁移能力增加(图6H),同时CXCL9/10上调(图6I)。在Kyn或Mtb处理后,SOCS3的表达与对照组相比显著增加,但Kyn处理或Mtb感染并没有改变AhR沉默巨噬细胞中SOCS3的表达或JAK1或JAK2的靶蛋白磷酸化(图6J、K)。当用AhR抑制剂SR-1预处理细胞时,Kyn或Mtb感染对SOCS3的增强作用被消除(图6L、M)。综上,Kyn-AhR启动SOCS3,从而调节JAK-STAT1信号通路以影响宿主免疫。

图6. AhR通过上调SOCS3调控JAK-STAT1通路
7. Trp-Kyn代谢抑制会导致在Mtb感染期间T 细胞过早地聚集到肺部
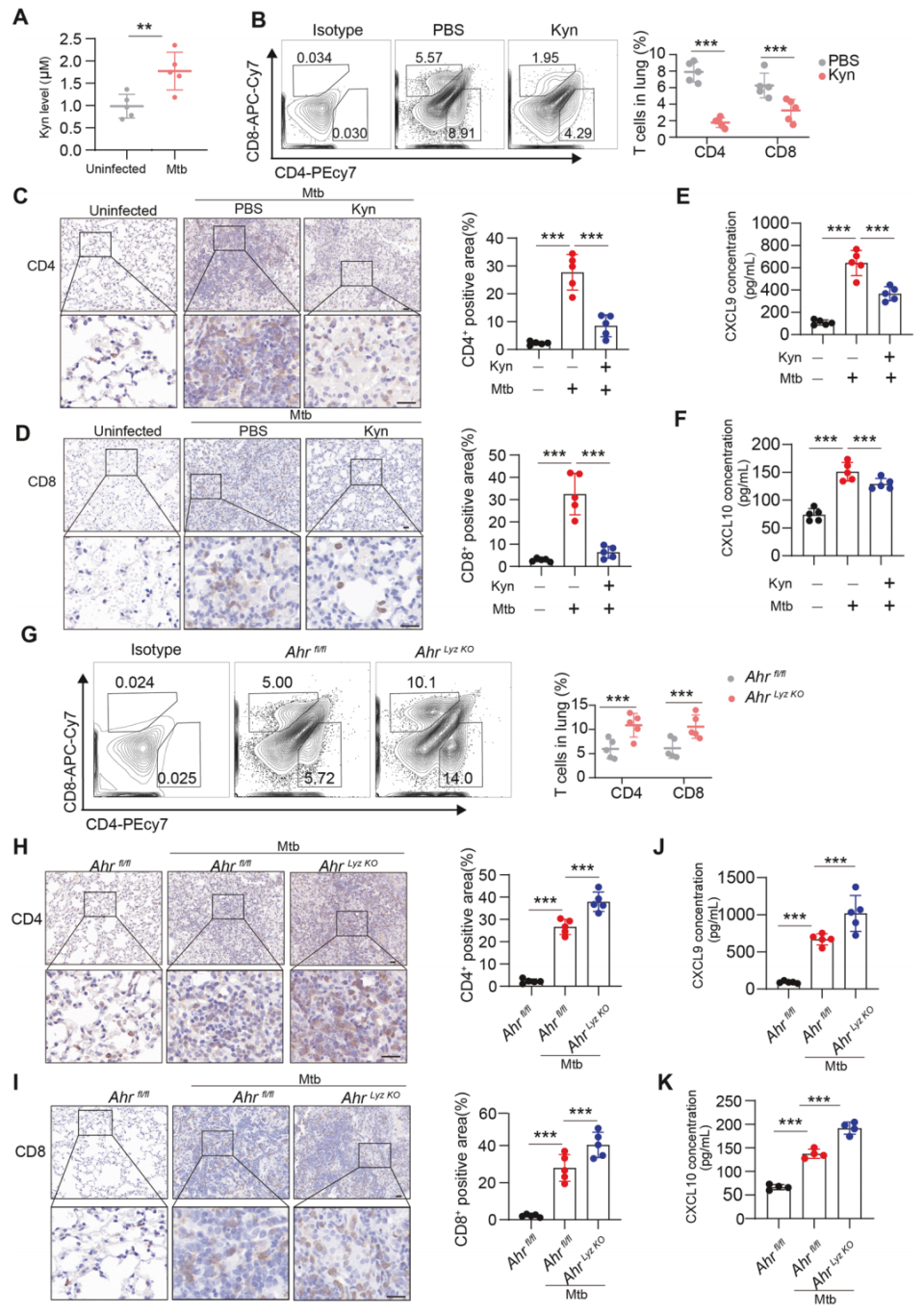
图7. Kyn-AhR通路削弱小鼠体内对Mtb感染的抵抗力
在AhrLyz-KO小鼠中,肺部CD4+和CD8+T细胞的浸润水平显著高于WT小鼠(图7G-I),且血清中CXCL9和CXCL10的表达水平升高(图7J、K)。前述实验表明,Mtb促进巨噬细胞中的Kyn-AhR信号传导,从而抑制体外T细胞浸润。后续研究进一步在小鼠模型中验证了该机制的体内作用。首先在小鼠模型中研究了Mtb感染期间Kyn-AhR信号通路对肺部T细胞的影响。尽管与未感染小鼠相比,Mtb感染小鼠的血清Kyn水平升高(图7A),但与PBS相比,Mtb感染小鼠的Kyn治疗导致肺CD4+和CD8+T细胞显著减少(图7B-D)。同样,血清中CXCL9和CXCL10的表达水平降低(图7E、F)。在AhrLyz-KO小鼠中,肺部CD4+和CD8+T细胞的浸润水平显著高于WT小鼠(图7G-I),且血清中CXCL9和CXCL10的表达水平升高(图7J、K)。
8. Kyn-AhR通路会导致CD4+T细胞功能障碍
将Kyn注射到Mtb感染的小鼠体内会导致抑制性受体上调(图8A、B),但会减少CD4+和CD8+T细胞释放效应分子IFN-γ和TNF-α(图8C、D)。与Ahrfl/fl小鼠相比,AhrLyz-KO小鼠肺CD4+和CD8+T细胞中免疫检查点标志物的表达水平较低(图8E、F),而IFN-γ和TNF-α的表达水平较高(图8G、H)。这些结果表明Kyn-AhR通路影响效应T细胞的功能。
将Kyn注射到Mtb感染的小鼠体内,以研究Trp代谢物Kyn对小鼠模型疾病负担和肺部病理学的影响。在野生型(WT)小鼠中,Kyn治疗显著增加了肺和脾中的细菌负荷(图8I、J)。CFU测定和抗酸染色表明,Ahr沉默增强了宿主针对Mtb的免疫反应,从而减少了肺部细菌负荷(图8K、L)。这些发现表明Kyn-Ahr通路抑制减轻了小鼠结核病模型的疾病负担。
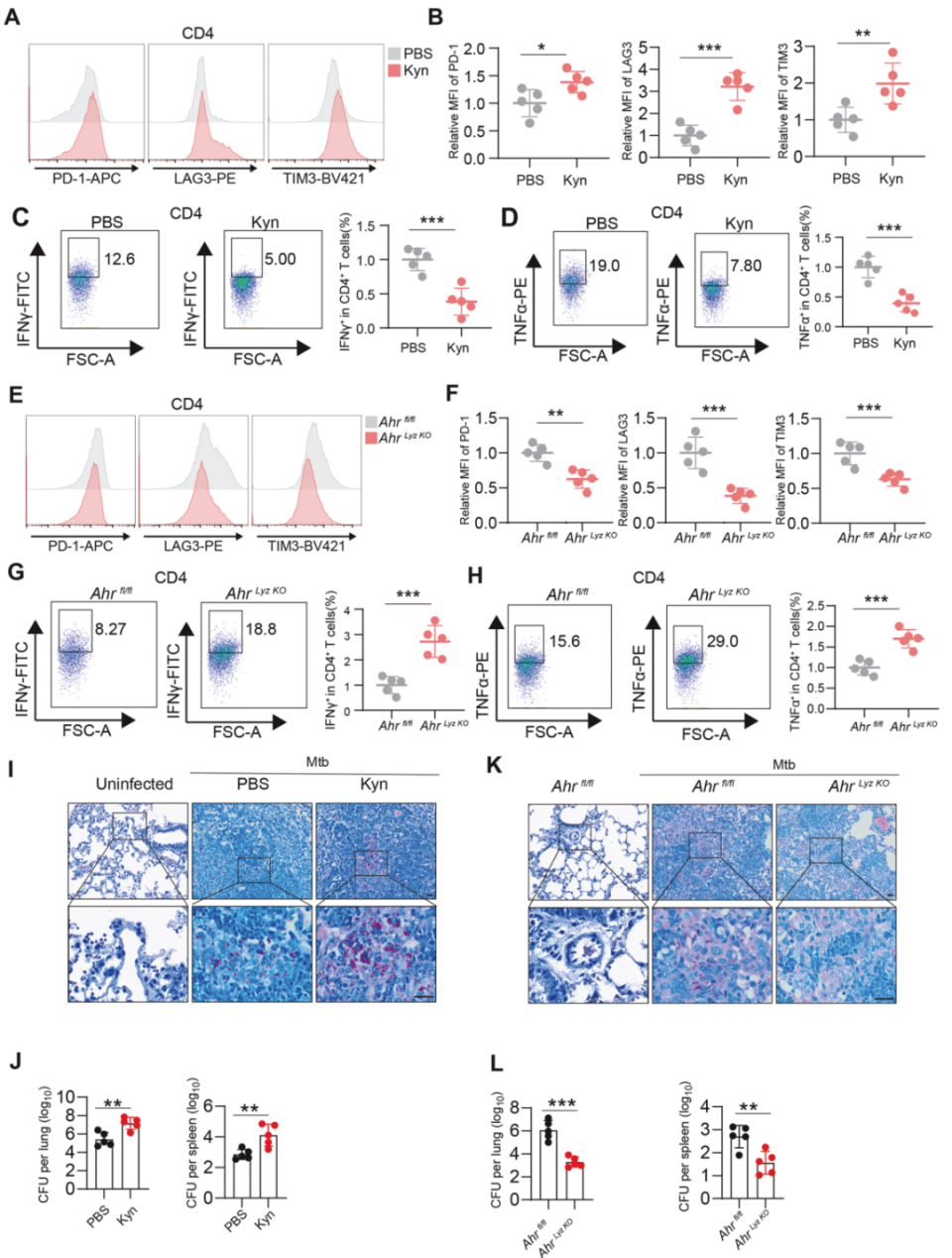
图8. Kyn-AhR通路导致CD4+T细胞功能障碍
结论
Mtb通过增加炎症巨噬细胞中IDO1的表达来促进Kyn的生成,进而增加细胞内AhR的表达。AhR随后上调SOCS3的表达,从而抑制JAK-STAT1通路的激活。这导致趋化因子表达减少,从而阻碍了T细胞向肺部感染部位的迁移。鉴于T细胞反应的延迟是治愈Mtb感染的主要障碍,也是新型结核病疫苗研发过程中的一个瓶颈,该研究结果为开发针对AhR的宿主导向治疗策略提供了重要的理论支持。抑制AhR的活性有助于加速T细胞的免疫反应,从而增强宿主对Mtb感染的控制能力,并为结核病的治疗和疫苗研发开辟新的途径。
研究点评
从临床视角看,当前结核病治疗虽对约95%的患者有效,但仍有约5%的病例(如危重症、骨结核、淋巴结结核等难治性肺外结核)面临治疗困境。传统抗菌疗法对这些患者往往效果有限,亟需免疫调节策略作为补充。本研究揭示的AhR免疫抑制通路,恰为这类难治性结核提供了新的干预思路:通过抑制AhR活性,可能解除其对T细胞招募与功能的抑制,从而加速免疫应答、增强感染控制。此外,通路中Trp向Kyn的代谢转化也是潜在干预环节,然而针对氨基酸代谢的广泛抑制在操作层面上面临挑战,且可能带来较大的脱靶风险。相比之下,更值得深入探索的是对该通路下游节点,如SOCS3蛋白,进行特异性干预。开发靶向SOCS3的小分子抑制剂或调控剂,有望更精准地解除其对JAK-STAT1通路的抑制,从而避免上游干预可能产生的系统性影响,这或许是更具转化潜力的研究方向。
新型免疫通路和标志物的发现不仅是结核病宿主导向治疗的重要靶点突破,也为难治性结核(尤其是肺外结核)的免疫辅助治疗开辟了潜在路径。未来,结合免疫疗法与现有抗结核化疗的联合方案,有望为那5%的“治疗困境”患者带来新的治愈希望,同时也为结核病治疗性疫苗或免疫佐剂的研发提供了关键的科学依据。
▌参考文献:
Liu X, Yang M, Xu P, et al. Kynurenine-AhR reduces T-cell infiltration and induces a delayed T-cell immune response by suppressing the STAT1-CXCL9/CXCL10 axis in tuberculosis. Cell Mol Immunol. 2024;21(12):1426-1440. doi:10.1038/s41423-024-01230-1

更多精彩内容,请扫码订阅“深三院结核之窗”专栏

卢水华 教授
教授,主任医师,二级教授,博士生导师
国家感染性疾病临床医学研究中心副主任,深圳市第三人民医院肺病医学部主任

陈珍妍
深圳市第三人民医院肺病一科博士后,四川大学卫生检验与检疫学士、复旦大学病原生物学博士
主要研究领域涉及结核病宿主免疫疗法及训练免疫,结核病诊断标志物筛选,抗结核感染免疫。累计发表学术论文10余篇,其中一作4篇,包括Int Immunopharmacol,Front Microbiol,Infection等 。曾任《结核病实验室检测与图解》编委,参与一项结核病诊断标志物相关专利,获结核病临床与检验高峰论坛(第四届)会议报告一等奖,参与多项国家级重点研发及自然科学基金项目,获批2026年度广东省自然科学基金,中国博士后科学基金第78批面上资助。
来源:《感染医线》
声 明
凡署名原创的文章版权属《感染医线》所有,欢迎分享、转载(开白可后台留言)。本文仅供医疗卫生专业人士了解最新医药资讯参考使用,不代表本平台观点。该等信息不能以任何方式取代专业的医疗指导,也不应被视为诊疗建议,如果该信息被用于资讯以外的目的,本站及作者不承担相关责任。
